![]()
|
|
|
|
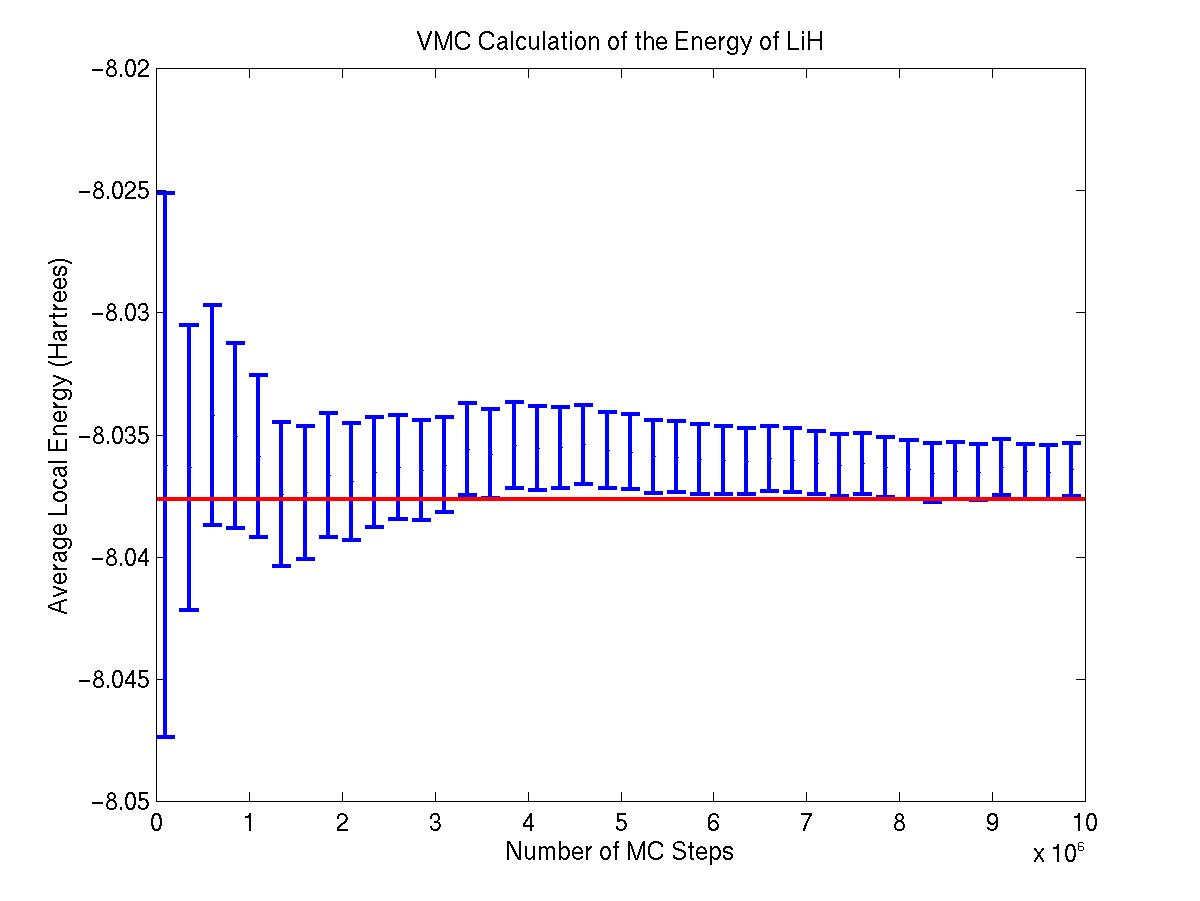
This page contains some preliminary results: Below is a variational Monte Carlo (VMC) calculation on LiH. The calculation ran for 10 million steps at an acceptance ratio of 0.5. The red line at -8.037 Hartrees demarcates Mark Dewing's result. Note that the graph represents the data from a SINGLE RUN, showing the convergence of the energy to the desired results. For this run, our code calculated the ground state energy of LiH to be -8.036 +/- 0.01 Hartrees.
Back to Top
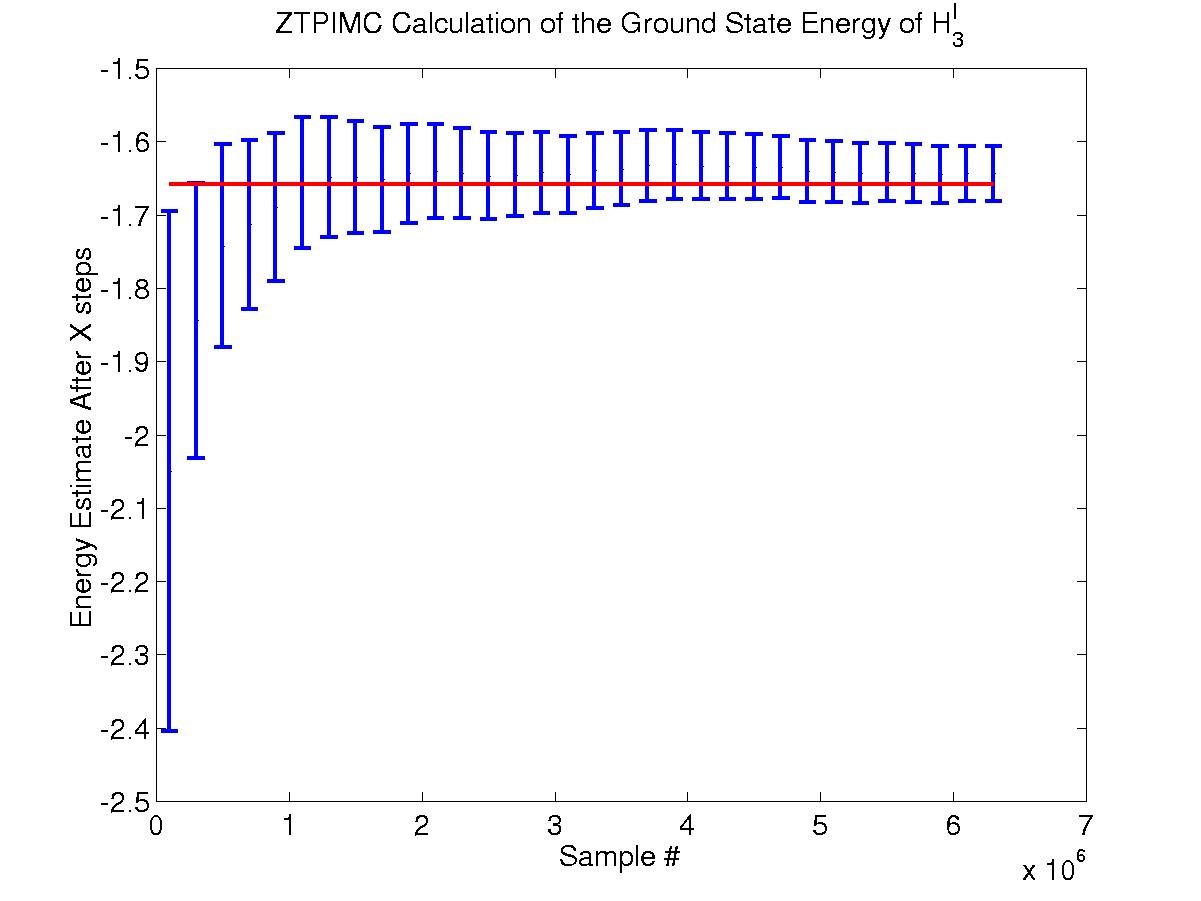
Below is a zero-temperature path integral Monte Carlo (ZTPIMC) on H3(I). The calculation ran for 6.5 million time steps at an acceptance ratio of 0.5. A time step of t=0.025 and m = 4 links was used. The red line at -1.6581(3) Hartrees is the fixed node calculation published in reference [1]. Note that the graph represents the data from a SINGLE RUN, showing the convergence of the energy to the desired results. From this run, our code calculated the ground state energy of H3(I) to be -1.642 +/- 0.1 Hartree. Note that the large error bars are a result of an extremely large autocorrelation time of approximately 2000 steps. This long autocorrelation results from the fact that we use only a simple displace move (in which each electron in each time slice is moved by a random amount in a random direction) to update our polymer chain. Thus, we do not have sufficiently refined statistics to ascertain the validity of our results.
Back to Top |
|
For problems or questions regarding this web contact nromero@uiuc.edu.
|