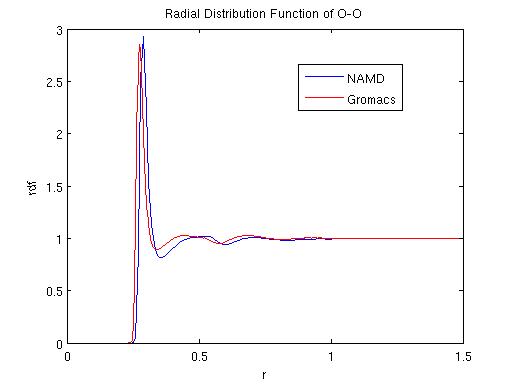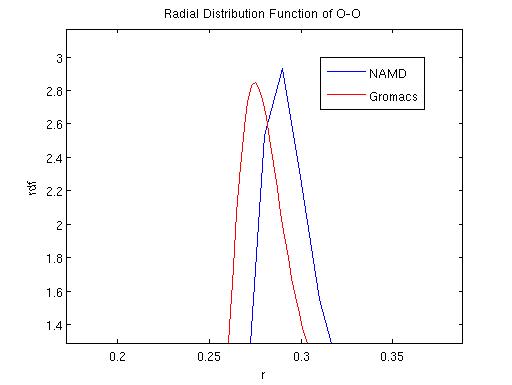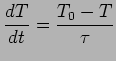
The two MD programs GROMACS (Lindahl et al., 2001) and NAMD (Phillips et al., 2005) are both widely used MD simulation packages with somewhat different emphasis: while GROMACS aims at fast simulations, NAMD is targeted at parallel computing of large systems. Many aspects of an MD simulation can cause different results using the two MD packages. For instance, the force fields used by the program are usually different. Other causes include long range potential calculation algorithm, e.g., PME or Ewards for electric interaction, basic simulation parameters such as time step, neighbor list cut-off radius, as well as temperature control scheme like Langevin, Berendsen or Nose-Hoover method. Usually SPC/E rigid water model (Berendsen et al., 1987) is utilized with Nose-Hoover or Berendsen temperature control scheme in GROMACS, while flexible TIP3P (Mahoney and Jorgensen, 2000) water model is commonly used with Langevin control scheme in NAMD.
Here we compare the two packages using a periodic system of water with and without ions. During our comparisons, we tried different combinations of all controllable parameters in the two packages. For details please refer to the appendix I. Our results indicate that using a same set of parameters, the two packages yield very similar results. Two different water models, SPCE and TIP3P were compared and it was found that the two models appear to have similar dynamic properties. We have also found that the damping coefficient in langevin dynamics may affect the simulation results dramatically and therefore has to be chosen with caution.
Below we describe the procedure of our simulations first, and then we present the results as well as analysis obtained from these simulations.
It is noteworthy that the force fields used by MD programs are often optimized at 300K. Therefore, in order to simulate our NVE ensemble at this temperature, a short (250 picosecond) ``warm-up'' simulation was performed to bring the system's temperature up to 300K. In order to eliminate any interference from the different temperature control schemes used by the two programs, this warm-up simulation was only carried out with NAMD. The final coordinates and velocities of the system was retrieved and used as a starting point for the microcanonical ensemble simulation.
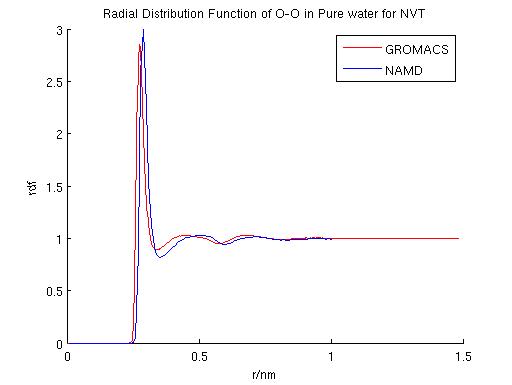
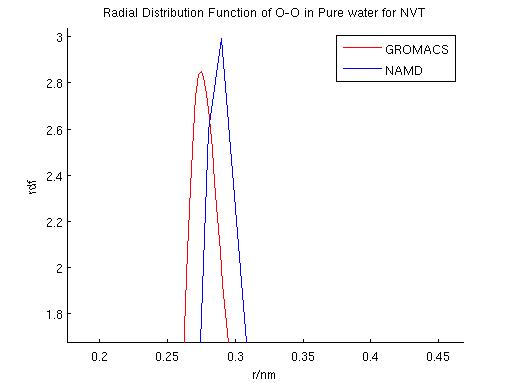
After the NVE simulation, a NVT ``zero point'' was also acquired using a similar procedure. Langevin dynamics was used in both GROMACS and NAMD in these simulations. The result for the NVT zero point simulations are shown in Table 2. According to the NVE and NVT simulations, diffusion coefficients calculated using the two packages are only 5.6% and 7.3% for NVE and NVT, respectively.
The Nose-Hoover algorithm extends the system's Hamiltonian by introducing a thermal reservoir and a friction term in the equations of motion. The friction force is proportional to the product of each particle's velocity and a friction parameter:
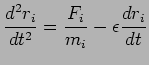
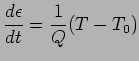
Compared with Nose-Hoover method, Langevin Dynamics introduces a random force, the equation of motion becomes:
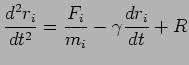
GROMACS and NAMD each adopts different temperature control schemes: GROMACS mainly uses Nose-Hoover and Berendsen thermostat while NAMD mainly uses Langevin dynamics or temperature coupling scheme. We performd NVT simulations using each of these temperature control schemes to compare their effects. Specifically, a first comparison was performed to evaluate the effect of damping coefficients in the simulation result using langevin dynamics. The fluctuation in total energy, temperature and radial distribution function were compared. Diffusion coefficients were also calculated and compared among the different schemes. A second comparison was performed among the three different temperature control schemes.
The effect of damping effects in Langevin Dynamics using TIP3P water model is summarized in Table. 3.
It can be seen from Table. 3 that damping affects the diffusion of water dramatically.
This can be interpreted from the equation of motion as shown above. The damping coefficient ![]() determines how much friction
an atom in the system will experience. The larger the
determines how much friction
an atom in the system will experience. The larger the ![]() is, the more friction the sytem will experience. If the langevin dynamics is applied
to water molecules, as in our case, you may picture a more and more ``sticky'' water when
is, the more friction the sytem will experience. If the langevin dynamics is applied
to water molecules, as in our case, you may picture a more and more ``sticky'' water when ![]() is getting larger.
In fact, if
is getting larger.
In fact, if ![]() is too large, the system will migrate from the inertia regime to the non-inertia regime, and obeys the rules
of Brownian dynamics (http://en.wikipedia.org/wiki/). Therefore,
is too large, the system will migrate from the inertia regime to the non-inertia regime, and obeys the rules
of Brownian dynamics (http://en.wikipedia.org/wiki/). Therefore, ![]() has to be chosen with caution if one's aim is to use Langevin dynamics to control the system temperature.
It is noteworty that the experimental value for water diffusion is
has to be chosen with caution if one's aim is to use Langevin dynamics to control the system temperature.
It is noteworty that the experimental value for water diffusion is
![]() , therefore,
using damping coefficient of 5/ps best reproduces the experimental result.
, therefore,
using damping coefficient of 5/ps best reproduces the experimental result.
For different temperature control scheme, we have the following three cases,
(1) Langevin dynamics with damping coefficient ![]() =5/ps
(2) Nose-hoover thermostat with
=5/ps
(2) Nose-hoover thermostat with ![]() =0.1 ps
(3) Berendsen thermostat with
=0.1 ps
(3) Berendsen thermostat with ![]() =0.1 ps
As shown in Table. 5, different temperature control schemes can achieve similar results with well-chosen parameters.
=0.1 ps
As shown in Table. 5, different temperature control schemes can achieve similar results with well-chosen parameters.
Although TIP3P is one of the most commonly used water models in MD simulations, various other water models are available, i.e., SPC, SPCE, TIP4P, TIP5P, etc. These different water models may each reproduce the experimental values of different properties of water. Therefore, we extended our simulations and compared the performance of two different water models, TIP3P and SPC. It can be seen from Table. 4 that the two models SPCE and TIP3P appear to have similar dynamic properties. At last, we move on to the water+ion system. As summarized in Table. 6 to Table. 8, for different force field, GROMACS and NAMD gave similar results.
| GROMACS | NAMD | |
| Water Modle | TIP3P | TIP3P |
| D (
|
4.9799 (+/- 0.0012) | 5.278 (+/-0.012) |
| GROMACS | NAMD | |
| Water Modle | TIP3P | TIP3P |
| Temperature control | Langevin ( |
Langevin ( |
| D (
|
2.5676 (+/-0.00009) | 2.769 (+/-0.002) |
| GROMACS | NAMD | |
| 4.0702 +/- 0.1798 | 4.259 +/- 0.025 | |
| 2.3815 +/- 0.1998 | 2.769 +/- 0.002 | |
| 1.9401 +/- 0.0403 | 1.958 +/- 0.003 |
| SPCE | TIP3P | |
| D (
|
2.3815 +/- 0.1998 | 2.5467 (+/- 0.0783) |
| D (
|
|
| Langevin | 2.5467 (+/- 0.0783) |
| Berendsen | 2.5454 (+/- 0.1814) |
| Nose-hooever | 2.4181 (+/- 0.2624) |
| Package | Temperature control scheme | Water model | D (
|
| NAMD | Langevin ( |
tip3p(namd)/charmm | 2.2247 (+/-0.0015) |
| GROMACS | Berendsen | spce (gromacs) | 2.3026 (+/- 0.00027) |
| GROMACS | Berendsen | spce (reference) | 2.4140 (+/- 0.00006) |
| GROMACS | Nose-Hoover | spce (gromacs) | 2.3235 (+/- 0.00024) |
| GROMACS | Nose-Hoover | spce (reference) | 2.4362 (+/- 0.00036) |
| Package | Temperature control scheme | Water model | D (
|
| NAMD | Langevin ( |
tip3p(namd)/charmm | 0.8200 (+/-0.0250) |
| GROMACS | Berendsen | spce (gromacs) | 0.9194 (+/- 0.00120) |
| GROMACS | Berendsen | spce (reference) | 1.3675 (+/- 0.00069) |
| GROMACS | Nose-Hoover | spce (gromacs) | 1.0353 (+/- 0.00021) |
| GROMACS | Nose-Hoover | spce (reference) | 1.2700 (+/- 0.00033) |
| Package | Temperature control scheme | Water model | D (
|
| NAMD | Langevin ( |
tip3p(namd)/charmm | 1.1125 (+/-0.0130) |
| GROMACS | Berendsen | spce (gromacs) | 1.3300 (+/- 0.00064) |
| GROMACS | Berendsen | spce (reference) | 1.6091 (+/- 0.00100) |
| GROMACS | Nose-Hoover | spce (gromacs) | 1.3517 (+/- 0.00050) |
| GROMACS | Nose-Hoover | spce (reference) | 1.4956 (+/- 0.00061) |
Simulation parameter
Water Box: 3X3X3 nm![]()
Number of Atoms: 2685
Density: 991.637 (g/l)
Time-step: 1fs
Electrostatic algorithm:
Long range: PME
Short range: cutoff at 1.0 nm
van der Waals interaction:
switch radius: 1.0 nm cutoff radius: 1.2 nm
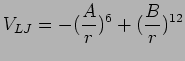
The extended simple point charge (SPC/E) model is characterized by three
point masses with O-H distance of 0.1 nm, H-O-H angle equal to 109.47![]() ,
charges on the oxygen and hydrogen equal to -0.8476 e and +0.4238 e,
respectively, and with Lennard-Jones parameters of oxygen-oxygen interaction
according to equation (11.2) (A = 0.37122 (kJ/mol)1/6· nm and B =
0.3428 (kJ/mol)1/12· nm) (Berendsen et al., 1987).
The SPC/E model has a dipole moment of 2.35 D. The diffusion
constant is improved considerably compared to the SPC model. The agreement
of the radial density distribution with experiment is somewhat better
for the SPC/E model than for the SPC model
(Karniadakis, G., Beskok, A., Aluru, N., Microflows and Nanoflows, Springer, 2005).
,
charges on the oxygen and hydrogen equal to -0.8476 e and +0.4238 e,
respectively, and with Lennard-Jones parameters of oxygen-oxygen interaction
according to equation (11.2) (A = 0.37122 (kJ/mol)1/6· nm and B =
0.3428 (kJ/mol)1/12· nm) (Berendsen et al., 1987).
The SPC/E model has a dipole moment of 2.35 D. The diffusion
constant is improved considerably compared to the SPC model. The agreement
of the radial density distribution with experiment is somewhat better
for the SPC/E model than for the SPC model
(Karniadakis, G., Beskok, A., Aluru, N., Microflows and Nanoflows, Springer, 2005).
H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem. 91 (1987) 6269-6271
Mahoney and Jorgensen (2000). J. Chem. Phys. 112, 8910-8922
Nose, S., A molecular dynamics method for simulations in the canonical ensemble, Mol. Phys., ver 52, pp255-268, 1984
Allen, M.P., Tildesley, D.J., Computer Simulations of Liquids, Oxford Science Publications, 1987
S. Koneshan, Jayendran C. Ransaiah, R. M. Lynden-Bell, S. H. Lee, Journal of Physics and Chemistry B, 1998, 102, 4193-4204
Daan Frenkel and Berend Smit. Understanding Molecular Simulations, Academic Press, 2nd edition, 2001.
This document was generated using the LaTeX2HTML translator Version 2002-2-1 (1.70)
Copyright © 1993, 1994, 1995, 1996,
Nikos Drakos,
Computer Based Learning Unit, University of Leeds.
Copyright © 1997, 1998, 1999,
Ross Moore,
Mathematics Department, Macquarie University, Sydney.
The command line arguments were:
latex2html -split 0 report
The translation was initiated by Yi Wang on 2007-05-14