Noble Gases
These are the non-bonding,
rare gases (e.g., Kr and Ar) where the electronic shells are closed.
Thus means they are spherically symmetric, with rather weak,
but long-ranged, van der Waals type interactions.
They were the first systems to be simulated and pair potentials
work best for them. For this case, a oft used pair potentials
is the Lennard-Jones (6-12) potential.
At large distances (r >> r0, the equilibrium distance)
one can undestand the interaction by considering how two oscillators
would interact, giving an r-6 attraction, where the
coefficient is determined by the atomic polarizibility (arising
due ot correlations between electron clouds surrounding atoms
and give van der Waals (dipole-dipole) interactions.) On the
other hand, at short distances (r << r0), the potentials
are strongly repulsive because of the Pauli exclusion principle
(electrons are fermions). Lennard-Jones assumed a stronger inverse
power law, conveniently r-12. One should not think
the Lennard-Jones (L-J) potential as anything more fundamental
than this.
Even for Argon it is only accurate to 10% or so.
And three-body potentials can be significant at the
10% level (more in case of polarizable species).
An example of the difference between the simple L-J 6-12
potential and one that has been constructed using a
large quantity of experimental data is on pg. 8 of A&T.
The potentials that are fit to bulk data are really
effective potentials and could be somewhat different
than the real 2-body potential that would be determined
from gas-phase data because they include corrections
for three- and higher-body interactions, for, in effect,
the two-body potential is obtained by averaging over the
third spatial coordinate:
(1/Volume) 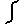 drk
V3i (ri,rj,rk)
= V2(3)(rij)
so that V(r)=(1/2)
drk
V3i (ri,rj,rk)
= V2(3)(rij)
so that V(r)=(1/2) ij
V2eff(rij).
ij
V2eff(rij).
Hence, the effective potentials can be dependent upon density and less
so on temperature.
Metals: Other problems with 2-body interactions
How about metals?
Metals are characterized by a rare gas shell plus a number of
valence electrons. Pair potentials do not work terribly well,
because the electrons will be delocalized.
If one does use a pair potential, it must be softer than the
Lennard-Jones form, for example a Morse potential (sum of exponentials.)
The Morse potential is often fit by choosing equilibrium lattice constant,
the bulk moduli, and the cohesive energy. This is the basis for the
so-called Rose equation of state and is the form used in
Effective Medium Theory (see below).
The major problem with pair potentials for solid metals is that they have
no environmental dependence so the a bulk atom is too much like a surface atom.
For metals, the strength of the individual bonds decrease as the local
environment becomes more crowded, corresponding to the behavior expected
from Pauli's "exclusion principle." In contrast, with 2-body potentials
the "strength" of the individual bonds does not depend on environment.
Thus, capturing the environmental dependence of bonding is crucial for
metallic systems.
Failure: An example of how
2-body interactions like L-J fail is the relaxation of a surface layer of atoms
in a metallic system, like Cu. Pair potentials predicted expansion of surface
layer, whereas, most metals contract inward to increase the charge
density and, therefore, the metallic bonding.
In the formula page we show the
potential used in the Embedded Atom Method (EAM) for metals.
The basic idea is that metallic bonding involves ions in an
electron sea so the total potential energy is the sum of direct
ion-ion interactions (e.g., to keep ion cores from overlapping)
and the interactions between ion and the electron sea. One assumes
that locally there is an electroni charge density coming
from the postive charge of the ion and nearby ions and that
the interaction is some function of this density. This latter
term is then a true many-body potential. The EAM works well
for spherically symmetric atoms (close-packed solids) such
as Cu, Al, and Pb.
References and other related concepts, derived from Quantum
Mechanics, are:
- Embedded Atom Method [EAM] -
Daw and Baskes, Phys. Rev. B 29, 6443 (1984);
Foiles, Baskes, and Daw, Phys. Rev. B 33, 7983 (1986)
- bond-order potential -
e.g., Horsfield, J. Phase Equil. 18, 573 (1997);
Aoki et al., ibid. 614; and applications, e.g.:
Lagana et al., J. Chem. Phys. 108, 3886 (1998);
Girshick et al, Phil. Mag. A77, 981 (1998)},
- Effective Medium Theory [EMT] -
Jacobsen, Norskov, Puska, Phys. Rev. B35, 7423 (1987).
- Finnis-Sinclair (FS)- Phil. Mag A50, 45 (1985);
Tersoff Phys. Rev. B37 6991 (1988).
- Glue Model - e.g., Ercolessi et al., Phil Mag. A58, 23 (1988)
These methods have very different parameterizations and functions.
In addition, while they usually appear composed of 2-body-type potentials,
all really are cleverly disguised MANY-body potentials.
Covalently-bonded Molecules
When molecules form bonds it is best to put that in directly.
Silicon has been studied extensively since it is so important for the
semiconductor industry. Silicon forms bonds with the nearest 4 atoms with
a local tetrahedron structure. This is very unlikely to ever come out of
a pair potential so it has to be put in.
In the formula page we give the well-known
potential function for silicon: the Stillinger-Weber potential.
It contains an explicit term which forces the angle between nearby triples to
stay close to 109 degrees and has been adjusted to giving roughly the lattice
constant, the cohesive energy, the melting point and liquid structure.
Unfortunately, this does not work well for other polytypes found under
pressure, e.g., for there is no bond coordination changes or bending
allowed: NO TRANSFERABILITY. There is a recent review article
Balamane, Phys. Rev. 46, 2250 (1992)
describing how well various potentials work on silicon.
They conclude that the potentials, even if they have been carefully tuned,
do not really describe all aspects of pure silicon terribly well. That is why
the Car-Parrinello technique is viewed as an advance (even though it is slow).
One gets a better (but still not perfect) potential, without have to adjust
lots of parameters.
For other systems there are extensive codes which handle real
biomolecular systems (GROMOS, CHARM, AMBER ...).
They use an experimental data base and put in specific terms for
bond lengths, bond angles torsion angles. etc.
They are really quite complicated because they have to distinguish
between many local bonding arrangements.
Even so, one should never trust the accuracy of the empirical potentials.
The errors can still be quite large even if a lot of data has been put in.
This is because the potential surface is so highly dimensional and there
are so many different possible combinations of atoms that experiment can
only hope to sample a very few of them. (See A&T pgs 6-24)
 E.
E.