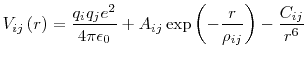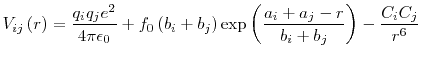Next: Molecular Dynamics Up: Development of Generalized KMC Previous: Introduction Contents
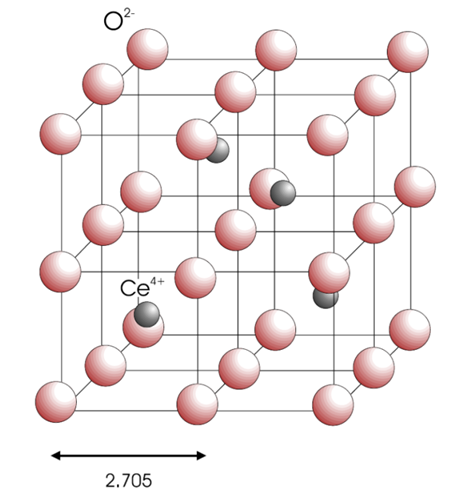
Fluorite structure oxides exhibit similar material properties, such as high radiation tolerance and high thermal stability. Cerium oxide is attractive as a surrogate for uranium oxide because it has the same fluorite crystal structure and many similar material properties, including melting temperature (UO
![]() , CeO
, CeO
![]() ) and thermal diffusivity, and has been well characterized experimentally up to
) and thermal diffusivity, and has been well characterized experimentally up to
![]() [2][3][4][5][6].
[2][3][4][5][6].
While ionic conductivity is believed to be negligible in pure ceria, it increases significantly when ceria is doped with an aliovalent oxide like Y![]() O
O![]() and La
and La![]() O
O![]() . The open structure of the fluorite lattice is able to tolerate the high level of atomic disorder that would be introduced by this type of doping. When ceria is doped with a trivalent ion like Lanthanum which forms La
. The open structure of the fluorite lattice is able to tolerate the high level of atomic disorder that would be introduced by this type of doping. When ceria is doped with a trivalent ion like Lanthanum which forms La![]() O
O![]() , a local charge imbalance is created. The lattice must compensate for this excess negative charge using one of three mechanisms: vacancy compensation, dopant interstitial compensation, and cerium interstitial compensation [7]. The mechanisms can be represented in Kröger-Vink notation:
, a local charge imbalance is created. The lattice must compensate for this excess negative charge using one of three mechanisms: vacancy compensation, dopant interstitial compensation, and cerium interstitial compensation [7]. The mechanisms can be represented in Kröger-Vink notation:
 |
Since its development in the 1970s, Molecular Dynamics has been widely used to predict various material properties. However, while it has been used extensively in metal and metal alloy systems, its use in ceramic oxide systems has been comparatively lacking. This is likely due to a lack of confidence in available interatomic potentials. While the potentials in metal and metal alloy systems have been well developed and validated by experimental results, the potentials for ceramic alloys generate results that are not always consistent with experimental data.
Since the the understanding of atomic level interactions is very important in nuclear fuel research, a series of studies have been done to model and understand the thermodynamics and defect chemistry of uranium oxide (UO![]() ) [9][10][11][12][13][14][15][16][17][18]. An extensive literature review was provided by Grovers et al. [19][20], where all of the available interatomic potentials on UO
) [9][10][11][12][13][14][15][16][17][18]. An extensive literature review was provided by Grovers et al. [19][20], where all of the available interatomic potentials on UO![]() were compared. They found that none of the potentials reviewed could predict all of the thermodynamic, mechanical, and defect transport properties. For example, they found that oxygen vacancy migration energies obtained from simulations ranged from
were compared. They found that none of the potentials reviewed could predict all of the thermodynamic, mechanical, and defect transport properties. For example, they found that oxygen vacancy migration energies obtained from simulations ranged from ![]() eV to
eV to ![]() eV, while the experimentally measured energy was found to be
eV, while the experimentally measured energy was found to be ![]() eV. Similarly, oxygen interstitial migration energies obtained from simulations ranged from
eV. Similarly, oxygen interstitial migration energies obtained from simulations ranged from ![]() eV to
eV to ![]() eV, while the experimentally measured energies ranged from
eV, while the experimentally measured energies ranged from ![]() eV to
eV to ![]() eV. These discrepancies could be attributed to the fact that all of the potentials reviewed were obtained by fitting parameters to experimentally measured properties, such as lattice constant, lattice energy, dielectric constants, etc. As most of these potentials were fitted without considering defect energetics, their inability to accurately predict these values is possibly unavoidable. This survey also demonstrated the importance of testing and validating potentials before using them for predictive purposes.
eV. These discrepancies could be attributed to the fact that all of the potentials reviewed were obtained by fitting parameters to experimentally measured properties, such as lattice constant, lattice energy, dielectric constants, etc. As most of these potentials were fitted without considering defect energetics, their inability to accurately predict these values is possibly unavoidable. This survey also demonstrated the importance of testing and validating potentials before using them for predictive purposes.
While there are nineteen interatomic potentials available for UO![]() , there are only a few potentials available for CeO
, there are only a few potentials available for CeO![]() , and even fewer with dopant parameters. Through an extensive literature survey conducted by Dr. Di Yun, only eight potentials for CeO
, and even fewer with dopant parameters. Through an extensive literature survey conducted by Dr. Di Yun, only eight potentials for CeO![]() were found. From those eight, three were selected for comparison in this study. Similar to what was found Grovers' literature review for UO
were found. From those eight, three were selected for comparison in this study. Similar to what was found Grovers' literature review for UO![]() [19][20], all of the potentials found for CeO
[19][20], all of the potentials found for CeO![]() were in two forms.
were in two forms.
The first potential form consists of the addition of a Buckingham term to the basic Coulomb potential. This form can be described by:
The second potential form consists of the addition of a Morse potential to the Buckingham potential form in Equation 2.5, which is used to describe the covalent bonding between the anions and cations. This form can be described by:
| Parameters | Units | Potentials | ||
| Gotte [22] | Minervini [7] | Sayle [23] | ||
| O shell charge | e | -6.5667 | -2.04 | -6.1 |
| O core charge | e | 4.5667 | 0.04 | 4.1 |
| O spring constant | eV/Å |
1759.8 | 6.3 | 419.9 |
| Ce shell charge | e | 4.6475 | 4.2 | 7.7 |
| Ce core charge | e | -0.6475 | -0.2 | -3.7 |
| Ce spring constant | eV/Å |
43.451 | 177.84 | 291.75 |
| O - O interactions | ||||
| A | eV | 9533.421 | 9547.96 | 22764.3 |
| Å | 0.234 | 0.2192 | 0.149 | |
| C | Å |
224.88 | 32 | 43.83 |
| O - Ce interactions | ||||
| A | eV | 755.1311 | 1809.68 | 1986.83 |
| Å | 0.429 | 0.3547 | 0.35107 | |
| C | Å |
0 | 20.4 | 20.4 |
| O - La interactions | ||||
| A | eV | 2088.79 | ||
| Å | 0.346 | |||
| C | Å |
23.25 | ||
Previous work has been done using both modeling and experimental techniques to investigate the clustering of oxygen vacancies around dopant ions in ceria doped with trivalent ions (in this system, the lanthanum trapping effect). Wang et al. showed that in the dilute range, charged dimers
![]() form [24]. Gerhardt-Anderson and Nowick extended this work on other
form [24]. Gerhardt-Anderson and Nowick extended this work on other
![]() , and suggested that conductivity and dimer binding energy vary inversely with dopant cation radius [25]. Kilner and Brook found that due to the size mismatch between the host and dopant cations, elastic strain energy makes a large contribution to the binding energy of the dimer [26]. Subsequent theoretical studies also showed the importance of defect clustering in the determination of free charge carrier concentration in fluorite oxides [27][28][29].
, and suggested that conductivity and dimer binding energy vary inversely with dopant cation radius [25]. Kilner and Brook found that due to the size mismatch between the host and dopant cations, elastic strain energy makes a large contribution to the binding energy of the dimer [26]. Subsequent theoretical studies also showed the importance of defect clustering in the determination of free charge carrier concentration in fluorite oxides [27][28][29].
Previous work has also been carried out to validate this type of local configuration dependent kinetic Monte Carlo technique. Murray et al. carried out one of the earliest investigations modeling oxygen vacancy conductivity in yttria-doped cerium oxide [30]. They showed that the oxygen vacancy barrier is sensitive to the local dopant environment and that a first nearest neighbor approximation could generate ionic conductivity results that are somewhat consistent with experimental results. Pornprasertsuk et al. used a similar KMC approach by calculating the binding energies of the oxygen vacancies and dopant ions in Yttria-stabilized zirconia [31]. They showed that the association energy of oxygen vacancies and dopant ions was big enough compared to the oxygen vacancy migration barrier to make the vacancy migration energy depend on the local dopant environment. These studies helped to validate the use of local configuration-dependent migration energies in kinetic Monte Carlo simulations, but these codes were written specifically for the systems in question. The goal of this study was to create a generalized code that could be used to simulate arbitrary systems with arbitrarily complex local configurations, so long as the migration energies for these configurations are known.
Aaron Oaks 2010-05-10